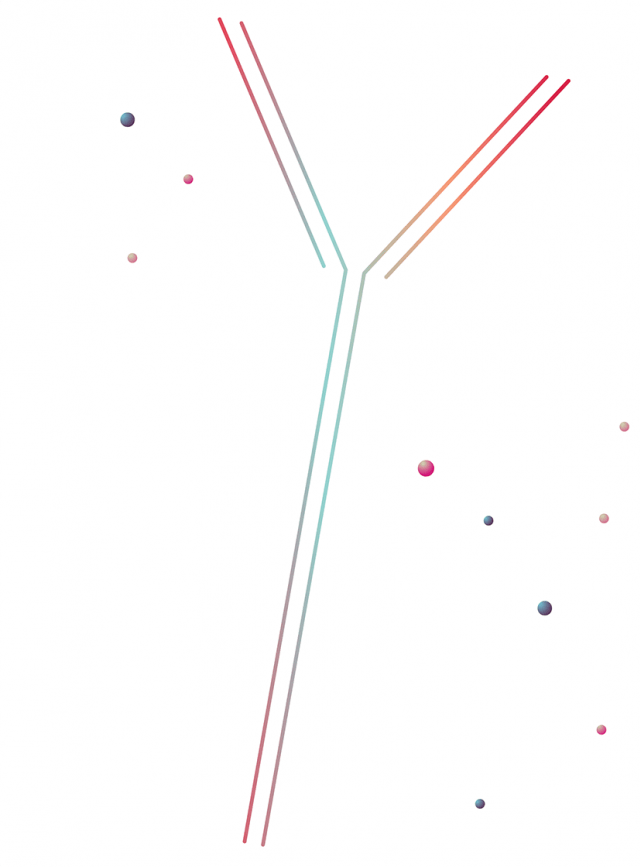One of the enduring mysteries of COVID-19 is why it makes some people deathly sick but gives others only mild symptoms or none at all. We know that age matters, as do race; gender; and pre-existing medical conditions like high blood pressure, heart problems, and obesity. We also know that most people who succumb to the disease die because they develop severe pneumonia, also known as acute respiratory distress syndrome, or ARDS.
What we still don’t know, however, is what tips one COVID-19 victim toward ARDS but not another. Early in the pandemic, doctors noticed that, compared to forms of ARDS caused by other respiratory infections like flu, some features of COVID ARDS were peculiar. Patients were not only slower to develop the syndrome but also slower to recover, in some cases spending weeks on a ventilator. Often, their immune systems continued a ruinous battle against their own bodies – ravaging their lungs and choking them of oxygen – even after SARS-CoV-2, the virus that causes COVID-19, had been cleared from their systems.
Image
Carolyn Calfee, MD, MAS ’09, critical care physician
“It was very strange,” says UC San Francisco’s Carolyn Calfee, MD, MAS ’09, a critical care physician and one of the world’s leading experts on ARDS. “I thought, ‘What kind of ARDS does this?’” she recalls. “‘This is not normal.’”
It turns out that a lot about COVID-19 is not normal. Experts have learned that in addition to infecting the respiratory tract, SARS-CoV-2 can infect the heart, gut, and blood vessels. As in the lungs, however, the damage that the virus inflicts on these tissues often appears to pale in comparison to the destruction caused by patients’ own immune response. Inflammation is rampant and widespread – turning up even in the brain and the toes – and causes myriad debilitating symptoms that sometimes persist for months. In rare cases, children (and some adults) who have recovered from COVID-19 develop a mysterious inflammatory syndrome, in which many organs throughout the body become inflamed.
All of this has piqued scientists’ curiosity: What is going wrong with people’s immune systems?
In searching for answers, Calfee and other researchers are finding that COVID-19 unhinges the immune system in ways no one expected, going so far as to turn the body against itself. Some people who get especially bad or unusual symptoms, for instance, harbor rogue antibodies – similar to those seen in autoimmune diseases – that disrupt the body’s normal immune response or attack its own tissues. These discoveries could explain how the virus wreaks such extensive and variable harm; they could also help predict who, if infected, will fall dangerously ill and identify effective treatments. More profoundly still, they could change scientists’ fundamental understanding of human immunity and how it can go awry.
The emerging insights into the immunology of COVID-19 could change scientists’ fundamental understanding of human immunity and how it can go awry.
A Broken Alarm
Alexis Combes, PhD, couldn’t sleep. An early-career scientist at UCSF, he had grown up in Marseille, France, where a childhood illness had sparked a reverence for medical science. He arrived in San Francisco in 2017 to train in clinical immunology and, less than two years later, was hired to direct a new, state-of-the-art laboratory for UCSF Bakar ImmunoX, an initiative that is exploring the immune system’s role in cancer and other diseases in hopes of inspiring treatments.
Image
Alexis Combes, PhD, director of D2B CoLab
In April 2020, members of ImmunoX teamed up with Calfee and other clinical researchers to launch a study examining how people’s immune systems respond to the new coronavirus. The study, called COMET (COVID-19 Multi-Phenotyping for Effective Therapies), required collecting blood and other bodily substances from hospitalized patients and then processing the samples – extracting fragile immune cells, genes, and other molecules – so that researchers could analyze them. Combes, whose lab specializes in this kind of deep investigation, jumped at the chance to help.
Still, he felt anxious. He lay awake, picturing himself pipetting tiny drops of blood from people dying of COVID-19, and hoped that nothing would go horribly wrong. “Later on, we learned there is almost no virus in the blood because it stays in the lungs,” Combes says, “but back then, we didn’t really know how dangerous these samples were.”
Around 4 a.m., he rose and dressed. The light-rail train he normally took into the lab had stopped running, so he rented an electric Lyft bicycle and pedaled the 15-minute commute on dark, abandoned streets. He arrived early so that he could get samples from the hospital’s morning blood draw while the specimens were still fresh. He had to work fast. Within hours, many important but short-lived immune cells would already be dead.
Combes ran the blood through a series of assays designed to pluck out immune cells and decode the genetic material in each cell, one by one – a process called single-cell sequencing. He wore two pairs of gloves, two lab coats, and an N95 mask. He focused on sequencing RNA, a marker of which genes (DNA) a cell has turned on. Like a computer’s task manager, which shows the software programs currently in use, the decoded RNA would reveal which genetic programs a patient’s immune cells were running in response to the coronavirus infection.
Together with a couple dozen technicians and trainees, Combes repeated this process of extracting and sequencing immune cells every morning for nearly two months. Then he and other COMET scientists began to dig into the resulting data.
Immune cells deploy a variety of programs for fighting viruses. One of the most powerful is known as the interferon response. Some immune cells are stationed along the body’s borders, such as in the skin or a nasal membrane. When one of these sentinel cells detects a virus attack, the cell emits warning proteins called interferons – the biochemical equivalent of an air raid siren. Nearby cells respond to the interferons by turning on antiviral genes, which slow the reproduction of the virus and induce any cells that do become infected to commit suicide.
Parsing the sequencing data, Combes’s team noticed that the immune cells from patients with mild COVID-19 (those discharged from the hospital within a few days) were running this crucial virus-defense program without a hitch. But that was not the case in the immune cells from patients with severe COVID-19 (those admitted to the ICU, often with ARDS). Alarmingly, none of their cells had deployed the interferon response.
Image
Max Krummel, PhD, chair of ImmunoX
“What’s concerning is this is something we see across the entire immune system,” says Matthew “Max” Krummel, PhD, the chair of ImmunoX, UCSF’s Smith Professor of Experimental Pathology, and co-lead of the COMET study. Without an interferon response to keep a virus contained, he explains, an invader is free to spread rapidly and widely. Increasing numbers of ambushed cells would call for reinforcements by flooding the bloodstream with inflammatory proteins called cytokines, setting off what’s known as a cytokine storm. Fresh troops arriving from the blood, such as white cells and antibodies, would then attempt to flush out the virus through biological carpet-bombing, resulting in extensive inflammation and tissue damage – much like that seen in COVID ARDS.
By fall 2020, scientists around the world had amassed similar evidence pointing to the same conclusion: In the sickest COVID-19 patients, something was shutting down their interferon response. But what?
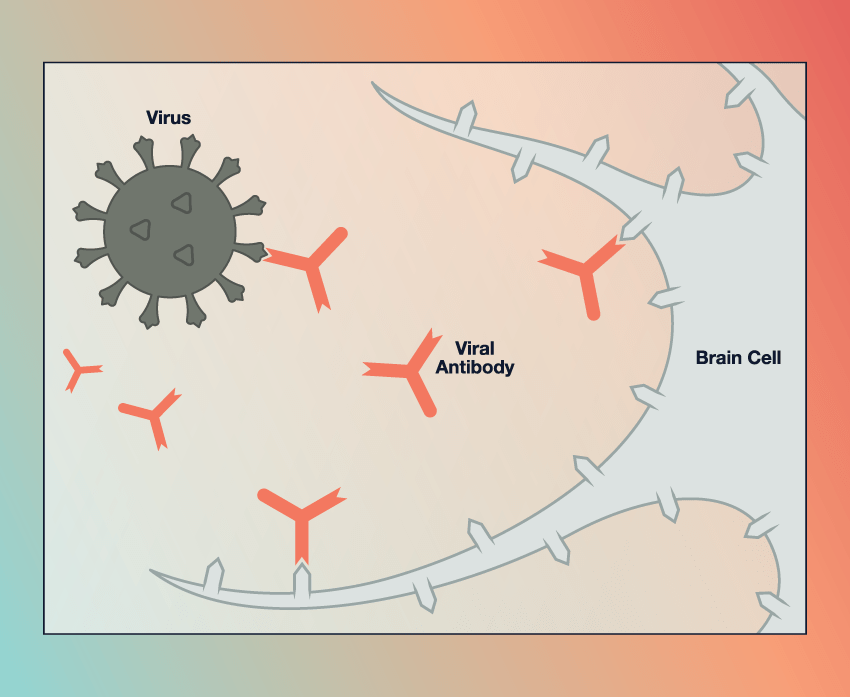
How Antibodies Thwart the Body’s Virus Alarm
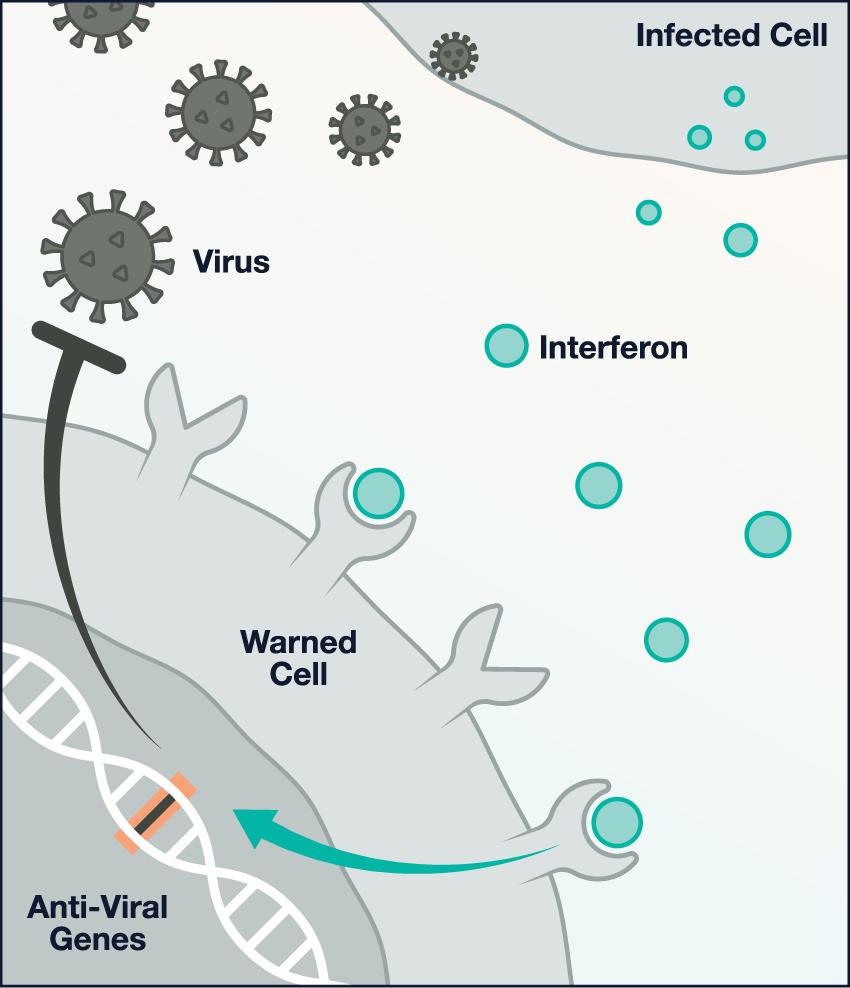
Image
A functional alarm: During a viral attack, infected cells emit warning proteins, called interferons, that cause nearby cells to turn on anti-viral genes. This interferon response prevents an infection from rapidly spreading.
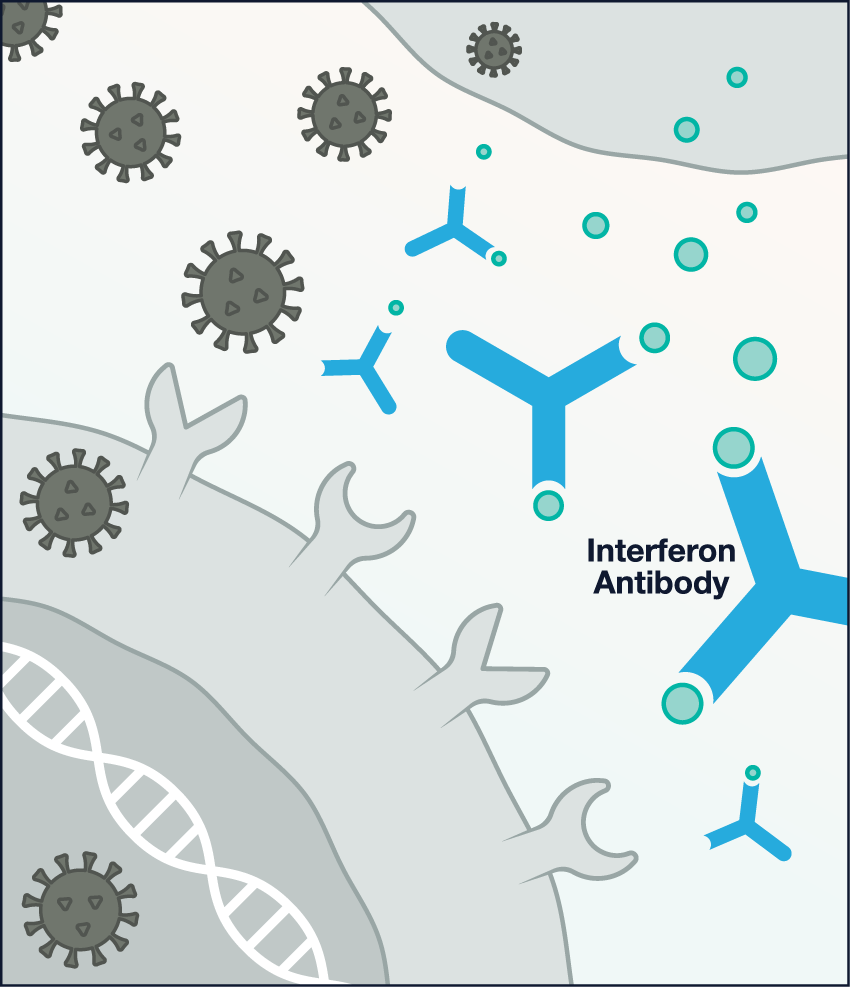
Image
Antibody attacks: About 10% of patients with severe COVID-19 have antibodies in their blood that attack interferons and thus prevent an interferon response.
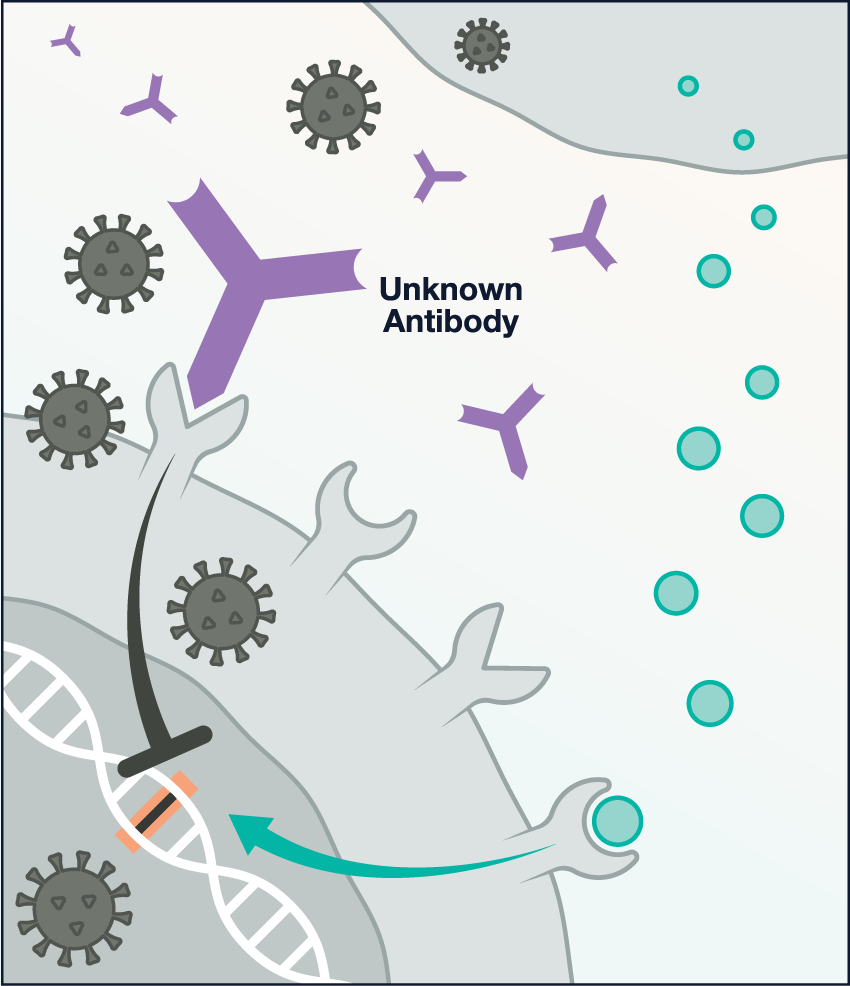
Image
Antibody interference: In other patients with severe COVID-19, a variety of (currently unknown) antibodies may bind to a cellular receptor called CD32B, which overrides the interferon signal. Illustrations: Chris Philpot
The Autoimmune Connection
One day in February 2020, a 32-year-old woman checked into a hospital in Lombardy, Italy. She was struggling to breathe and had a fever and a cough. Her symptoms, no surprise, were caused by COVID-19. Because she was young, her doctors thought she would recover quickly. But her condition worsened. She remained in the hospital for 37 days, including a week on a ventilator. By the time she was discharged, the burgeoning coronavirus outbreak had sickened more than 85,000 people in Italy. Her case might have been lost in the wave, save for one odd detail: She happened to have a rare autoimmune disease called autoimmune polyglandular syndrome type 1, or APS1.
Image
Mark Anderson, MD, PhD, co-founder of ImmunoX
APS1 is unusual in that it’s linked to heritable mutations in a single gene; these mutations cause an astonishing array of autoimmune conditions, including type 1 diabetes, endocrine dysfunction, and chronic fungal infections. Mark Anderson, MD, PhD, a co-founder of ImmunoX and UCSF’s Friend Professor of Diabetes Research, has studied APS1 for more than 20 years. The miscreant gene, he and others have learned, plays a role in teaching the immune system to ignore, or tolerate, the body’s own cells and molecules. In APS1, self-tolerance breaks down, permitting the immune system to generate renegade warrior cells and antibodies that make civilians and allies the targets of war. One of these ally targets is the immune system’s virus alarm protein, interferon.
Antibodies that target interferons capture these proteins the way a glove traps a baseball – stopping them from delivering their warning and tagging them for destruction by immune cells. These antibodies are so abundant in APS1 patients that their presence is often used to diagnose the disease. Weirdly, though, they had never seemed to do much harm, Anderson notes, since patients had tended not to get bad cases of the flu and other viral infections. “Then, lo and behold, COVID comes along, and word gets out on the street that a few APS1 patients who got the virus had a very poor clinical course,” he says. After the Italian case, two more popped up in Maryland.
Image
The implication – that hidden autoimmunity could turn an otherwise benign infection deadly – was almost too crazy to believe.
Then, in early August, Anderson got a call from Jean-Laurent Casanova, MD, PhD, an immunologist at Rockefeller University in New York City. Upon examining blood from nearly 1,000 people hospitalized with life-threatening COVID-19, Casanova told Anderson, his team had been shocked to find that more than 100 of them (about 1 in 10 people) had antibodies against interferons, just like the APS1 patients. None of these COVID patients, however, had any known autoimmune diseases.
In laboratory experiments, Casanova’s team showed that these antibodies block the interferon response in cells exposed to SARS-CoV-2, thus preventing the cells from resisting the virus and controlling its spread. The team had found no such antibodies in people with mild or asymptomatic COVID-19. They appeared to be a feature – a cause, likely – of some of the worst outcomes.
Image
Sara Vazquez, MD-PhD student
The implication – that hidden autoimmunity could turn an otherwise benign infection deadly – was almost too crazy to believe. “When I heard about it, I was like ‘Oh, my goodness, I have to see if it’s true because this is totally paradigm-shifting,’” says Sara Vazquez, an MD-PhD student who works with Anderson and Joe DeRisi, PhD, UCSF’s Gordon Tomkins Professor and an infectious-disease expert.
Immunologists, Vazquez points out, have long recognized a relationship between autoimmunity and infectious disease. Autoimmune diseases frequently follow microbial attacks, which can trigger these diseases in people with genetic and other predispositions. Streptococcus pyogenes, the species of bacterium behind strep throat, for example, is thought to cause rheumatic heart disease when antibodies generated to fight the bacterium attack tissues in the heart. However, hints that rogue antibodies could change the course of a viral infection were few and far between.
Image
Jimmie Ye, PhD, systems biologist
Intrigued, Vazquez decided to repeat the Rockefeller experiment in blood samples from COMET patients and others at Zuckerberg San Francisco General Hospital. “I’m still amazed we were able to replicate it,” she says. The Rockefeller data, which the journal Sciencepublished in October, hadn’t been a fluke: In San Francisco, as in New York, antibodies against interferons lurked in the blood of roughly 10% of patients brought to the brink of death or killed by COVID-19. Working with UCSF systems biologist Jimmie Ye, PhD, Vazquez and her team showed that immune cells from these patients had failed to run the interferon-response program, implying that the antibodies were directly responsible for the patients’ bad COVID outcomes. (For unknown reasons, almost everyone who had interferon antibodies was male, perhaps providing a clue as to why more men than women have died from COVID-19.)
What had caused these antibodies to appear is a mystery. One possibility is the coronavirus itself, but Vazquez and her colleagues doubt that’s the culprit. Patients with interferon antibodies showed consistently high levels of them throughout their hospital stays, she explains, “meaning those people probably had the antibodies before they ever got COVID.” She also tested blood from more than 4,000 people living in San Francisco’s Mission District who had never been infected with SARS-CoV-2 and found that 13 of them nevertheless had interferon antibodies.
If the ratio revealed by the Mission study holds up, something on the order of 10 million people worldwide could have these antibodies. They may be more likely to become very sick if they get COVID-19. “We’re dealing with an unusual pathogen,” Anderson says. “It’s now so rampant, it’s plucking people out of the general population who don’t even know they have this susceptibility,” which he speculates may result from a combination of genetics and previous viral infections. “Their ability to respond against the virus is slowed down, and the virus starts winning.”
Antibodies Gone Awry
Image
Illustration: Radhika Patnala Sci-Illustrate
Interferon antibodies may be just the tip of the iceberg. Their discovery raised an obvious question: If these antibodies are responsible for around 10% of severe COVID-19 cases, what accounts for the other 90%?
Combes’s and Krummel’s research points to one plausibility. In January, their team published findings in Nature showing that a variety of other, unknown antibodies from severe COVID-19 patients can also quash the interferon response without directly capturing interferons. To grasp how this works, it’s helpful to understand the relationship between antibodies and immune cells.
An antibody, which is shaped like a Y, has essentially two parts. The “arms” of the Y grab a specific target, such as a piece of a virus or, in unfortunate cases, an interferon. The “stem” of the Y, meanwhile, connects to a protein receptor on the surface of immune cells, like a plug in a socket. In this way, an antibody delivers instructions to an immune cell according to the type of receptor its stem plugs into. A typical message is: “I found the enemy – attack!” The Nature study revealed that antibodies from severely ill COVID-19 patients deliver to cells almost the opposite refrain: “Ignore the air-raid sirens.”
The receptor that receives this command goes by the name CD32B. Among other functions, Krummel thinks, it serves as a brake on the immune system once an infection is under control. Normally, this is a good thing. During a virus attack, he explains, cells running the interferon-response program shut down their operating machinery in order to keep the virus from hijacking it. “If your cells did that forever, you’d die,” he says. “CD32B is a way for antibodies to say to a cell, ‘We’re here. We’ve come to the rescue. Go ahead and get back to business because you’ve got to live.’” But in severe COVID-19, antibodies may deliver this message erroneously, perhaps communicating it too soon or too strongly. As a result, the antibodies turn off the interferon response while it’s still needed – in effect, pitting the immune system against itself.
Scientists are still exploring what these signal-jamming antibodies are and why they’re made. Krummel suspects that many of them are autoimmune, meaning that their “arms” attack the body’s own molecules – and not just interferons. He points to a study from Yale in which researchers engineered yeast cells to display some 3,000 human proteins. In exposing the cells to antibodies from COVID-19 patients, the researchers found that these antibodies targeted a wide variety of proteins. These included signaling proteins like interferons as well as proteins commonly expressed in cells and tissues throughout the human body. The worse a patient’s infection, the more kinds of human proteins their antibodies targeted.
“That’s super weird, because you’d think antibodies are supposed to be the good guys, right?” Krummel asks. “They’re supposed to protect you, not make you sicker.”
Immunologists have a theory, called the “danger model,” that could explain how a coronavirus infection might induce the immune system to churn out self-attacking antibodies, also known as autoantibodies. According to this theory, the immune system does not distinguish between self and non-self but rather between things that are dangerous and things that are not. Such an approach would seem to enable the immune system to root out not just viruses and other pathogens but also diseased cells, such as cancers.
A novel virus that the body has never seen before, Krummel speculates, might evoke a cacophony of danger signals that beckon and mobilize immune cells over many days. Caught up in the enduring commotion, some human proteins might get mistakenly labeled as “dangerous,” causing the immune system to make antibodies against them. Inflammation at the site of the infection or in other tissues might then recruit these autoantibodies while also prompting the immune system to generate more of them, resulting in a runaway autoimmune reaction. The inflammation that sets off this reaction could be caused by the invading virus itself or an unrelated infection or trauma.
Inflammation is also a hallmark of aging. “As people get older, they get more inflammatory cues in their blood, as if they’re responding to something all the time,” Krummel says. The same is true with obesity, he points out. A link between inflammation and autoantibodies might explain why COVID-19 disproportionately sickens both the elderly and the overweight.
Attacks on the Brain
Researchers at UCSF’s Weill Institute for Neurosciences have begun to suspect that autoantibodies might also have a hand in COVID-19’s strange smorgasbord of neurological symptoms. About a third of people hospitalized for the disease exhibit these symptoms, including headaches, muscle fatigue, impaired cognition (or “brain fog”), and loss of the senses of smell and taste. For some people, including those who experience only mild respiratory symptoms, brain and behavior problems can persist or develop even after they recover. For instance, patients are more likely to receive a new psychiatric diagnosis, such as anxiety disorder or dementia, after a bout with COVID-19 than after some other health event.
Image
Michael Wilson, MD ’07, MAS ’16, associate professor of neurology
A few months into the pandemic, experts started to wonder if SARS-CoV-2 was infecting some people’s brains. Would researchers find traces of the virus in their cerebrospinal fluid, which bathes the brain and spinal cord? “Almost universally, the answer was ‘no,’” says Michael Wilson, MD ’07, MAS ’16, an associate professor of neurology at UCSF’s Weill Institute. He knew, however, that brain-infecting viruses, such as West Nile, often show up in cerebrospinal fluid fleetingly – for just a day or two – even when patients are sick for months, making these viruses easy to miss. Consequently, physicians diagnose West Nile by testing for viral antibodies, which persist in cerebrospinal fluid much longer.
Image
Christopher Bartley, MD, PhD, postdoctoral fellow in immunopsychiatry
So Wilson, who is also the University’s Rachleff Distinguished Professor, teamed up with other scientists at UCSF and Yale to look for antibodies in samples of cerebrospinal fluid from hospitalized COVID-19 patients with neurological symptoms. “That we would find anything seemed very far-fetched,” says psychiatrist Christopher Bartley, MD, PhD, a postdoctoral fellow in immunopsychiatry at the Weill Institute, who co-led the experiments in the labs of Wilson and of Samuel Pleasure, MD, PhD, UCSF’s Johnson Professor of Neurology. But to the team’s surprise, the first seven fluid samples they tested all contained antibodies against SARS-CoV-2.
Image
Colin Zamecnik, PhD, bioengineer
“That’s very unusual,” says team member Colin Zamecnik, PhD, a UCSF bioengineer who is building new tools to screen for antibodies, including previously unknown ones. “The brain is compartmentalized from the rest of your immune system,” he explains. “The antibodies that you have circulating in your blood” – to fight an infection in the lungs, say – “don’t just passively show up in cerebrospinal fluid.”
Even more curious was the fact that five of the seven fluid samples also contained autoantibodies. These antibodies bind to brain tissue in mice, Bartley says, “which means they probably bind to brain tissue in humans.” The team found some brain-targeting antibodies in cerebrospinal fluid from uninfected people, too, but they appeared less frequently and proved to latch onto mouse brain tissue less capably than did those in the fluid from the hospitalized COVID-19 patients.
Image
Mimicry trickery: In rare cases, some people might produce antibodies against a coronavirus protein that resembles a protein in brain tissue, thereby triggering an immune attack on the brain. UCSF scientists are investigating whether this theory, known as molecular mimicry, could help explain COVID-19’s strange array of neurological symptoms. Illustration: Chris Philpot
Intriguingly, Bartley and his colleagues also discovered brain autoantibodies in three patients who developed acute psychiatric symptoms after having otherwise mild or asymptomatic COVID-19. One patient, a 30-year-old man with no history of mental illness, became suddenly violent and developed delusions that he was speaking to the dead and being experimented on “like a guinea pig.” He recovered after being treated with immunotherapy. When Bartley’s team analyzed the autoantibodies in his cerebrospinal fluid, they found that some of them targeted brain cells thought to be involved in schizophrenia.
Precisely how autoantibodies relate to COVID-19 and sufferers’ symptoms is unclear, although Pleasure’s lab is developing a mouse model to try to unpack this relationship. “The clinical story is still murky,” Wilson says. “Could antibody attacks be behind some of this weird neuro stuff? That’s a definite possibility, but we don’t have enough data yet to know.”
He suspects that, ironically, some patients might produce brain autoantibodies to battle the coronavirus itself. These antibodies may target a part of the virus that, by chance, resembles a part of a protein in brain tissue. This phenomenon is known as molecular mimicry. “It’s guilt by association,” Wilson explains.
His team is now taking a closer look at the coronavirus antibodies from COVID-19 patients to see if some of those same antibodies also target brain proteins – evidence of molecular mimicry at play. So far, they have identified two antibodies in one patient that seem to fit the bill. “It might just be some weird fluke,” Wilson says. “But if it turns out to have legs, it could be an interesting wrinkle.”
Cures for COVID and Beyond
The more we learn about antibodies and their effect on COVID-19, the better we may be able to identify who is likely to fall badly ill and how to treat them. Anderson, the APS1 specialist, is now working with colleagues in the UCSF Department of Laboratory Medicine to develop a clinical test for interferon antibodies. “If it were up to me and money was no expense, anybody who comes into the hospital would get this test,” he says. “Probably only like one in 1,000 patients would test positive, but it could make a huge difference in outcomes because then we could intervene.”
For instance, patients with interferon or other immune-dysregulating antibodies might benefit from treatment with experimental antiviral or immune-based therapies early in their infection, says Calfee, the ARDS expert. “If we knew who those people are who are at risk for progression to serious disease, we could focus on getting them into clinical trials,” she says. Krummel, meanwhile, has co-founded a startup aimed at developing drugs that can restore the interferon response by barring antibodies from plugging into the immune-cell receptor that turns it off.
Emerging insights into the immunology of COVID-19 likely will have an impact beyond the current pandemic as well. “What we’re finding probably is not unique to COVID,” says ImmunoX’s Combes. In recent experiments not yet published, his team found that antibodies also appear to shut down the interferon response in many patients with bad cases of flu. If current or newly developed immunotherapies prove effective in preventing severe COVID-19, these therapies might help curb suffering and deaths from other viral diseases, including future outbreaks. (COVID-19 cases aside, ARDS kills more than 70,000 people in the U.S. every year.)
Likewise, knowledge of antibodies’ role in COVID-19’s neurological symptoms could have profound implications for psychiatry. Bartley points out that about 1 in 100 people have a psychotic disorder for which the cause is unknown. He suspects that brain autoantibodies triggered by viruses might be culpable in a small percentage of those cases. “COVID gave us the opportunity to ask: Is there an association between a viral infection and the onset of psychiatric symptoms?” he says. “We think the answer is yes.” If that’s also true for viruses other than SARS-CoV-2, he speculates, “the impact could be enormous.” Many patients who don’t respond to antipsychotics, for example, might be cured by immunotherapy.
“Suddenly there are so many new questions scientists are beginning to ask about autoimmunity and its relationship to infectious disease,” says Vazquez, the MD-PhD student. “It has opened up this whole new field that didn’t exist before.”